Next: Scattering pair calculations Up: Usage Previous: Usage Contents
The software contains pre-calculated atomic presets for ELWF(a-files), so that the changes in the following parameters will not be required. The meanings of the atomic calculation parameters are the following:
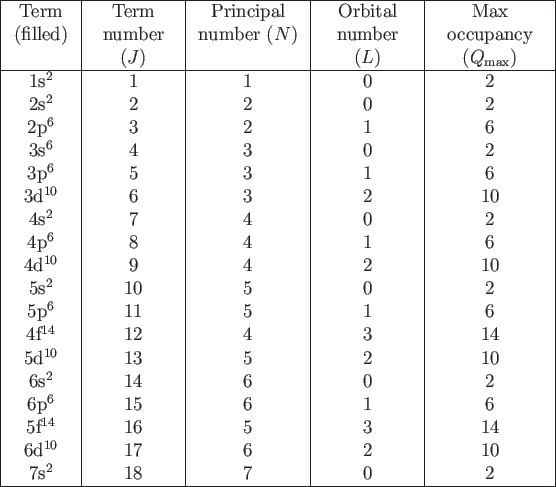
By default all terms up to ![]() -th are filled with electrons.
In order to simulate particular atom one should
change several (usually one) last terms
by setting appropriate set of parameters
-th are filled with electrons.
In order to simulate particular atom one should
change several (usually one) last terms
by setting appropriate set of parameters ![]() ,
, ![]() ,
, ![]() ,
,
![]() ,
, ![]() .
.
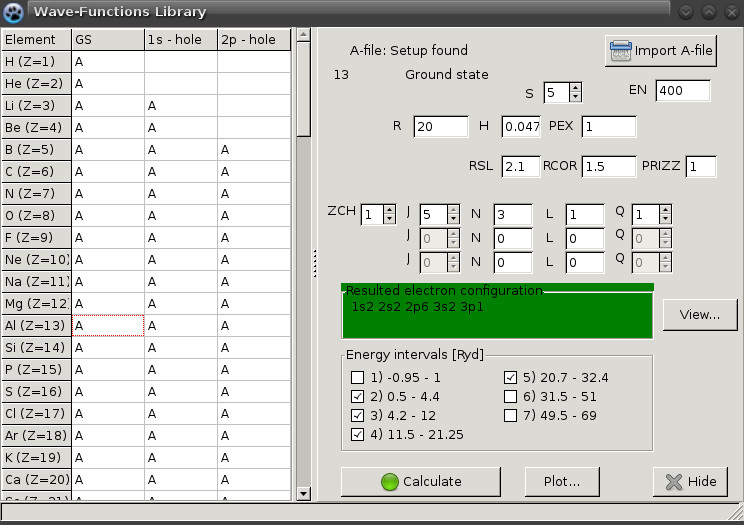
For example for simulation of Oxygen atom
in ground state
one should change the occupancy of 2p term
from default 2p![]() to actual 2p
to actual 2p![]() by
setting
by
setting ![]() ,
, ![]() ,
, ![]() ,
, ![]() in the atom wave function form shown on Figure 4.
in the atom wave function form shown on Figure 4.
In order to get atom with core hole
which should correspond to
the absorbing atom
one should change
1s (K-edge) or 2p (L
![]() 2,3-edge) term in the same way.
2,3-edge) term in the same way.
These are a bit annoying and error-rich parts of the calculation and we are working to make presets for all stable atoms to simplify user experience.
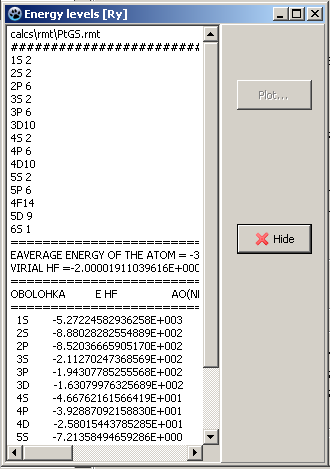
The resulting electron configuration could be verified by clicking View button. Window showed on Figure 5 should appear. The first data block here demonstrates accounted electron orbitals and their occupancy while the second shows energy levels of these orbitals (Ry).
The next important quantity is
![]() SL
SL![]() MT
radii.
It should be chosen at the value of total
potential
MT
radii.
It should be chosen at the value of total
potential
![]() C
C![]() XC = -1.1 Ry.
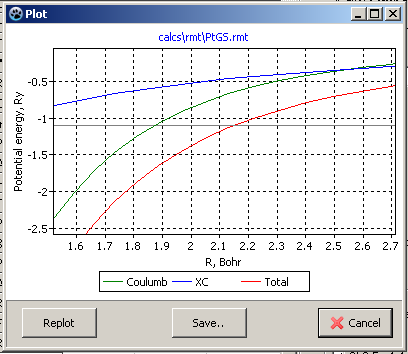
According the Figure 6,
XC = -1.1 Ry.
According the Figure 6,
![]() SL=2.2 Bohr for ground state
calculation of Platinum atom.
SL=2.2 Bohr for ground state
calculation of Platinum atom.
After these preparations all WFs may be computed. Just mark boxes corresponding to the calculated energy intervals and press Calculate button. Here ELWF code will be executed several times and B-files should appear in calc/b directory.
These calculation should be completed both for absorbing X-ray atom and his neighbors.
leon 2015-12-15